We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
The Survival Scatter plot shows the clinical status (i.e. dead or alive) for all individuals in the patient cohort, based on the same data that underlies the corresponding Kaplan-Meier plots. Patients that are alive at last time for follow-up are shown in blue and patients who have died during the study are shown in red.
The x-axis shows the expression levels (FPKM) of the investigated gene in the tumor tissue at the time of diagnosis. The y-axis shows the follow-up time after diagnosis (years). Both axes are complimented with kernel density curves demonstrating the data density over the axes. The top density plot shows the expression levels (FPKM) distribution among dead (red) and alive patients (blue). The right density plot shows the data density of the survived years of dead patients with high and low expression levels respectively, stratified using the cutoff indicated by the vertical dashed line through the Survival Scatter plot. This cutoff is automatically defined based on the FPKM cutoff that minimizes the p-score. The cutoff can be changed by dragging the vertical line or by entering a cutoff value in the square labeled "Current cut-off".
Under the Survival Scatter plot the p-score landscape (black curve; left axis) is shown together with dead median separation (red curve; right axis). Dead median separation is the difference in median mRNA expression between patients who have died with high and low expression, respectively. It is calculated as follows: median FPKM expression of dead patients with high expression - median FPKM expression of dead patients with low expression. This is intended to aid the user in visually exploring custom cutoffs and the associated p-scores and dead median separation.
Individual patient data is displayed and can be filtered by clicking on one or more of the category buttons on the top of the page. Categories describing expression level and patient information include: high, low, alive, dead, female, male and tumor stages. The scale of the x-axis can be toggled between linear and log-scale by clicking on the "x log" button. Mouse-over function shows TCGA ID, patient information and mRNA expression (FPKM) for each patient.
& Survival analysisi
Kaplan-Meier plots summarize results from analysis of correlation between mRNA expression level and patient survival. Patients were divided based on level of expression into one of the two groups "low" (under cut off) or "high" (over cut off). X-axis shows time for survival (years) and y-axis shows the probability of survival, where 1.0 corresponds to 100 percent.
The Survival Scatter plot shows the clinical status (i.e. dead or alive) for all individuals in the patient cohort, based on the same data that underlies the corresponding Kaplan-Meier plots. Patients that are alive at last time for follow-up are shown in blue and patients who have died during the study are shown in red.
The x-axis shows the expression levels (FPKM) of the investigated gene in the tumor tissue at the time of diagnosis. The y-axis shows the follow-up time after diagnosis (years). Both axes are complimented with kernel density curves demonstrating the data density over the axes. The top density plot shows the expression levels (FPKM) distribution among dead (red) and alive patients (blue). The right density plot shows the data density of the survived years of dead patients with high and low expression levels respectively, stratified using the cutoff indicated by the vertical dashed line through the Survival Scatter plot. This cutoff is automatically defined based on the FPKM cutoff that minimizes the p-score. The cutoff can be changed by dragging the vertical line or by entering a cutoff value in the square labeled "Current cut-off".
Under the Survival Scatter plot the p-score landscape (black curve; left axis) is shown together with dead median separation (red curve; right axis). Dead median separation is the difference in median mRNA expression between patients who have died with high and low expression, respectively. It is calculated as follows: median FPKM expression of dead patients with high expression - median FPKM expression of dead patients with low expression. This is intended to aid the user in visually exploring custom cutoffs and the associated p-scores and dead median separation.
Individual patient data is displayed and can be filtered by clicking on one or more of the category buttons on the top of the page. Categories describing expression level and patient information include: high, low, alive, dead, female, male and tumor stages. The scale of the x-axis can be toggled between linear and log-scale by clicking on the "x log" button. Mouse-over function shows TCGA ID, patient information and mRNA expression (FPKM) for each patient.
& Survival analysisi
Kaplan-Meier plots summarize results from analysis of correlation between mRNA expression level and patient survival. Patients were divided based on level of expression into one of the two groups "low" (under cut off) or "high" (over cut off). X-axis shows time for survival (years) and y-axis shows the probability of survival, where 1.0 corresponds to 100 percent.
The Survival Scatter plot shows the clinical status (i.e. dead or alive) for all individuals in the patient cohort, based on the same data that underlies the corresponding Kaplan-Meier plots. Patients that are alive at last time for follow-up are shown in blue and patients who have died during the study are shown in red.
The x-axis shows the expression levels (FPKM) of the investigated gene in the tumor tissue at the time of diagnosis. The y-axis shows the follow-up time after diagnosis (years). Both axes are complimented with kernel density curves demonstrating the data density over the axes. The top density plot shows the expression levels (FPKM) distribution among dead (red) and alive patients (blue). The right density plot shows the data density of the survived years of dead patients with high and low expression levels respectively, stratified using the cutoff indicated by the vertical dashed line through the Survival Scatter plot. This cutoff is automatically defined based on the FPKM cutoff that minimizes the p-score. The cutoff can be changed by dragging the vertical line or by entering a cutoff value in the square labeled "Current cut-off".
Under the Survival Scatter plot the p-score landscape (black curve; left axis) is shown together with dead median separation (red curve; right axis). Dead median separation is the difference in median mRNA expression between patients who have died with high and low expression, respectively. It is calculated as follows: median FPKM expression of dead patients with high expression - median FPKM expression of dead patients with low expression. This is intended to aid the user in visually exploring custom cutoffs and the associated p-scores and dead median separation.
Individual patient data is displayed and can be filtered by clicking on one or more of the category buttons on the top of the page. Categories describing expression level and patient information include: high, low, alive, dead, female, male and tumor stages. The scale of the x-axis can be toggled between linear and log-scale by clicking on the "x log" button. Mouse-over function shows TCGA ID, patient information and mRNA expression (FPKM) for each patient.
& Survival analysisi
Kaplan-Meier plots summarize results from analysis of correlation between mRNA expression level and patient survival. Patients were divided based on level of expression into one of the two groups "low" (under cut off) or "high" (over cut off). X-axis shows time for survival (years) and y-axis shows the probability of survival, where 1.0 corresponds to 100 percent.
The Survival Scatter plot shows the clinical status (i.e. dead or alive) for all individuals in the patient cohort, based on the same data that underlies the corresponding Kaplan-Meier plots. Patients that are alive at last time for follow-up are shown in blue and patients who have died during the study are shown in red.
The x-axis shows the expression levels (FPKM) of the investigated gene in the tumor tissue at the time of diagnosis. The y-axis shows the follow-up time after diagnosis (years). Both axes are complimented with kernel density curves demonstrating the data density over the axes. The top density plot shows the expression levels (FPKM) distribution among dead (red) and alive patients (blue). The right density plot shows the data density of the survived years of dead patients with high and low expression levels respectively, stratified using the cutoff indicated by the vertical dashed line through the Survival Scatter plot. This cutoff is automatically defined based on the FPKM cutoff that minimizes the p-score. The cutoff can be changed by dragging the vertical line or by entering a cutoff value in the square labeled "Current cut-off".
Under the Survival Scatter plot the p-score landscape (black curve; left axis) is shown together with dead median separation (red curve; right axis). Dead median separation is the difference in median mRNA expression between patients who have died with high and low expression, respectively. It is calculated as follows: median FPKM expression of dead patients with high expression - median FPKM expression of dead patients with low expression. This is intended to aid the user in visually exploring custom cutoffs and the associated p-scores and dead median separation.
Individual patient data is displayed and can be filtered by clicking on one or more of the category buttons on the top of the page. Categories describing expression level and patient information include: high, low, alive, dead, female, male and tumor stages. The scale of the x-axis can be toggled between linear and log-scale by clicking on the "x log" button. Mouse-over function shows TCGA ID, patient information and mRNA expression (FPKM) for each patient.
& Survival analysisi
Kaplan-Meier plots summarize results from analysis of correlation between mRNA expression level and patient survival. Patients were divided based on level of expression into one of the two groups "low" (under cut off) or "high" (over cut off). X-axis shows time for survival (years) and y-axis shows the probability of survival, where 1.0 corresponds to 100 percent.
Survival analysis data not available.
RENAL CELL CARCINOMA - Protein relative expression (CPTAC)
Number of samples
194
Samples
Sample ID
Sample type
nRPX
CPT0024670003
Tumor
2.2
CPT0025050003
Tumor
1.8
CPT0009000003
Tumor
1.7
CPT0086950003
Tumor
1.4
CPT0020120003
Tumor
1.3
CPT0001540009
Tumor
1.3
CPT0025110003
Tumor
1.2
CPT0012670003
Tumor
1.1
CPT0002350011
Tumor
1.1
CPT0081880003
Tumor
1.0
CPT0000780007
Tumor
1.0
CPT0092290003
Tumor
0.9
CPT0019990003
Tumor
0.9
CPT0088690003
Tumor
0.9
CPT0086030003
Tumor
0.9
CPT0001500009
Tumor
0.9
CPT0025230003
Tumor
0.8
CPT0079270003
Tumor
0.8
CPT0000640003
Tumor
0.8
CPT0012080003
Tumor
0.8
CPT0078510003
Tumor
0.8
CPT0025880003
Tumor
0.7
CPT0023350003
Tumor
0.7
CPT0015730003
Tumor
0.7
CPT0014450004
Tumor
0.7
CPT0064370003
Tumor
0.6
CPT0015810003
Tumor
0.6
CPT0063630003
Tumor
0.6
CPT0014160003
Tumor
0.6
CPT0009060003
Tumor
0.6
CPT0002270011
Tumor
0.6
CPT0006440003
Tumor
0.5
CPT0012280003
Tumor
0.5
CPT0079480003
Tumor
0.5
CPT0010110003
Tumor
0.5
CPT0007320003
Tumor
0.5
CPT0092160003
Tumor
0.5
CPT0069160003
Tumor
0.5
CPT0010160003
Tumor
0.4
CPT0001260009
Tumor
0.4
CPT0081600003
Tumor
0.4
CPT0065430003
Tumor
0.3
CPT0065690003
Tumor
0.3
CPT0012900004
Tumor
0.3
CPT0075560003
Tumor
0.3
CPT0086820003
Tumor
0.2
CPT0086360003
Tumor
0.2
CPT0088480003
Tumor
0.2
CPT0071150004
Tumor
0.2
CPT0079410003
Tumor
0.2
CPT0017410003
Tumor
0.2
CPT0069000003
Tumor
0.1
CPT0075720003
Tumor
0.1
CPT0088900003
Tumor
0.1
CPT0084590001
Normal
0.1
CPT0089460004
Tumor
0.1
CPT0012180003
Tumor
0.1
CPT0077110003
Tumor
0.0
CPT0065930003
Tumor
0.0
CPT0066470004
Tumor
0.0
CPT0088970003
Tumor
0.0
CPT0007860003
Tumor
0.0
CPT0026410003
Tumor
0.0
CPT0088550004
Tumor
0.0
CPT0012550003
Tumor
0.0
CPT0075130003
Tumor
0.0
CPT0078990003
Tumor
-0.1
CPT0092730003
Tumor
-0.1
CPT0001190001
Normal
-0.1
CPT0001340003
Tumor
-0.1
CPT0079230003
Tumor
-0.1
CPT0006630003
Tumor
-0.1
CPT0021240003
Tumor
-0.1
CPT0086870003
Tumor
-0.1
CPT0082010001
Normal
-0.1
CPT0085670003
Tumor
-0.1
CPT0009080003
Normal
-0.2
CPT0077140001
Normal
-0.2
CPT0077310003
Tumor
-0.2
CPT0017450001
Normal
-0.2
CPT0077490003
Tumor
-0.2
CPT0079380003
Tumor
-0.2
CPT0015910003
Tumor
-0.2
CPT0076350001
Normal
-0.2
CPT0089020003
Tumor
-0.2
CPT0075570001
Normal
-0.2
CPT0078530001
Normal
-0.2
CPT0001220008
Tumor
-0.2
CPT0088630003
Tumor
-0.2
CPT0013790003
Normal
-0.2
CPT0087040003
Tumor
-0.2
CPT0000870016
Tumor
-0.3
CPT0011240003
Tumor
-0.3
CPT0012770003
Normal
-0.3
CPT0078800003
Tumor
-0.3
CPT0012090003
Normal
-0.3
CPT0000890001
Normal
-0.3
CPT0063320003
Tumor
-0.3
CPT0076330003
Tumor
-0.3
CPT0018250001
Normal
-0.3
CPT0025610001
Normal
-0.3
CPT0065750003
Tumor
-0.3
CPT0012290003
Normal
-0.3
CPT0019130003
Tumor
-0.3
CPT0078670001
Normal
-0.3
CPT0088760003
Tumor
-0.3
CPT0025290003
Tumor
-0.3
CPT0065870003
Tumor
-0.4
CPT0001180009
Tumor
-0.4
CPT0006900003
Tumor
-0.4
CPT0026420001
Normal
-0.4
CPT0065450001
Normal
-0.4
CPT0014130001
Normal
-0.4
CPT0020020001
Normal
-0.4
CPT0092790003
Tumor
-0.4
CPT0012370003
Tumor
-0.4
CPT0014350001
Normal
-0.4
CPT0001350001
Normal
-0.4
CPT0006730001
Normal
-0.4
CPT0078930003
Tumor
-0.4
CPT0020130001
Normal
-0.4
CPT0009020003
Normal
-0.4
CPT0025060001
Normal
-0.4
CPT0092190003
Normal
-0.4
CPT0010170001
Normal
-0.4
CPT0012570003
Normal
-0.4
CPT0066480003
Tumor
-0.4
CPT0088710001
Normal
-0.4
CPT0001270001
Normal
-0.4
CPT0077320001
Normal
-0.4
CPT0077500001
Normal
-0.4
CPT0075170001
Normal
-0.4
CPT0088570001
Normal
-0.5
CPT0069010001
Normal
-0.5
CPT0089040001
Normal
-0.5
CPT0024680001
Normal
-0.5
CPT0088920001
Normal
-0.5
CPT0006950001
Normal
-0.5
CPT0087050003
Normal
-0.5
CPT0025170003
Tumor
-0.5
CPT0078840001
Normal
-0.5
CPT0064400001
Normal
-0.5
CPT0007870001
Normal
-0.5
CPT0066430001
Normal
-0.5
CPT0079430001
Normal
-0.5
CPT0088780001
Normal
-0.5
CPT0069190001
Normal
-0.5
CPT0012920003
Normal
-0.5
CPT0000790001
Normal
-0.5
CPT0092310003
Normal
-0.5
CPT0025920001
Normal
-0.5
CPT0081620001
Normal
-0.5
CPT0006530001
Normal
-0.5
CPT0063640001
Normal
-0.5
CPT0007470001
Normal
-0.6
CPT0092740003
Normal
-0.6
CPT0078940001
Normal
-0.6
CPT0014470001
Normal
-0.6
CPT0023710001
Normal
-0.6
CPT0086370003
Normal
-0.6
CPT0066520001
Normal
-0.6
CPT0079000001
Normal
-0.6
CPT0081990003
Tumor
-0.6
CPT0065820001
Normal
-0.6
CPT0001550001
Normal
-0.6
CPT0063330001
Normal
-0.6
CPT0023360001
Normal
-0.6
CPT0014370004
Tumor
-0.7
CPT0010120001
Normal
-0.7
CPT0019160001
Normal
-0.7
CPT0017850003
Tumor
-0.7
CPT0065810003
Tumor
-0.7
CPT0089480003
Normal
-0.7
CPT0023690003
Tumor
-0.7
CPT0086890003
Normal
-0.7
CPT0086970003
Normal
-0.7
CPT0011410003
Tumor
-0.8
CPT0002370001
Normal
-0.8
CPT0084560003
Tumor
-0.8
CPT0092800003
Normal
-0.8
CPT0086830003
Normal
-0.8
CPT0025350003
Tumor
-0.8
CPT0001510001
Normal
-0.8
CPT0001230001
Normal
-0.9
CPT0079300001
Normal
-0.9
CPT0078830003
Tumor
-0.9
CPT0012640003
Normal
-0.9
CPT0000660001
Normal
-0.9
CPT0088500001
Normal
-0.9
CPT0088640003
Normal
-0.9
CPT0079510001
Normal
-1.0
CPT0079180003
Tumor
-1.1
CPT0025580004
Tumor
-1.2
CPT0078660003
Tumor
-1.3
Show allShow less
RENAL CANCER - Protein expressioni
A mouse-over function shows sample information and annotation data. Click on an image to view it in a full screen mode. Samples can be filtered based on level of antibody staining by selecting one or several of the following categories: high, medium, low and not detected. The assay and annotation is described here.
Note that samples used for immunohistochemistry by the Human Protein Atlas do not correspond to samples in the TCGA dataset.
Antibody stainingi
Antibody staining in the annotated cell types in the current human tissue is reported as not detected, low, medium, or high, based on conventional immunohistochemistry profiling in selected tissues. This score is based on the combination of the staining intensity and fraction of stained cells.
Each image is clickable and will lead to virtual microscopy that enables deeper exploration of all samples and also displays staining intensity scores, fraction scores and subcellular localization as well as patient and tissue information for each sample.
 The Human Protein Atlas project is funded
The Human Protein Atlas project is funded





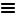



<br><b>Adenocarcinoma, NOS</b> (M-81403)<br><br><b>Antibody staining:</b> High<br><b>Intensity:</b> Strong<br><b>Quantity:</b> >75%<br><b>Location:</b> Cytoplasmic/membranous")
<br><b>Adenocarcinoma, NOS</b> (M-81403)<br><br><b>Antibody staining:</b> High<br><b>Intensity:</b> Strong<br><b>Quantity:</b> >75%<br><b>Location:</b> Cytoplasmic/membranous")
<br><b>Adenocarcinoma, NOS</b> (M-81403)<br><br><b>Antibody staining:</b> High<br><b>Intensity:</b> Strong<br><b>Quantity:</b> >75%<br><b>Location:</b> Cytoplasmic/membranous")
<br><b>Adenocarcinoma, NOS</b> (M-81403)<br><br><b>Antibody staining:</b> High<br><b>Intensity:</b> Strong<br><b>Quantity:</b> >75%<br><b>Location:</b> Cytoplasmic/membranous")
<br><b>Adenocarcinoma, NOS</b> (M-81403)<br><br><b>Antibody staining:</b> High<br><b>Intensity:</b> Strong<br><b>Quantity:</b> >75%<br><b>Location:</b> Cytoplasmic/membranous")
<br><b>Adenocarcinoma, NOS</b> (M-81403)<br><br><b>Antibody staining:</b> High<br><b>Intensity:</b> Strong<br><b>Quantity:</b> >75%<br><b>Location:</b> Cytoplasmic/membranous")
<br><b>Adenocarcinoma, NOS</b> (M-81403)<br><br><b>Antibody staining:</b> High<br><b>Intensity:</b> Strong<br><b>Quantity:</b> >75%<br><b>Location:</b> Cytoplasmic/membranous")
<br><b>Adenocarcinoma, NOS</b> (M-81403)<br><br><b>Antibody staining:</b> High<br><b>Intensity:</b> Strong<br><b>Quantity:</b> >75%<br><b>Location:</b> Cytoplasmic/membranous")
<br><b>Adenocarcinoma, NOS</b> (M-81403)<br><br><b>Antibody staining:</b> High<br><b>Intensity:</b> Strong<br><b>Quantity:</b> >75%<br><b>Location:</b> Cytoplasmic/membranous")
<br><b>Adenocarcinoma, NOS</b> (M-81403)<br><br><b>Antibody staining:</b> High<br><b>Intensity:</b> Strong<br><b>Quantity:</b> >75%<br><b>Location:</b> Cytoplasmic/membranous")
<br><b>Adenocarcinoma, NOS</b> (M-81403)<br><br><b>Antibody staining:</b> High<br><b>Intensity:</b> Strong<br><b>Quantity:</b> >75%<br><b>Location:</b> Cytoplasmic/membranous")
<br><b>Adenocarcinoma, NOS</b> (M-81403)<br><br><b>Antibody staining:</b> High<br><b>Intensity:</b> Strong<br><b>Quantity:</b> >75%<br><b>Location:</b> Cytoplasmic/membranous")
<br><b>Adenocarcinoma, NOS</b> (M-81403)<br><br><b>Antibody staining:</b> High<br><b>Intensity:</b> Strong<br><b>Quantity:</b> >75%<br><b>Location:</b> Cytoplasmic/membranous")
<br><b>Adenocarcinoma, NOS</b> (M-81403)<br><br><b>Antibody staining:</b> High<br><b>Intensity:</b> Strong<br><b>Quantity:</b> >75%<br><b>Location:</b> Cytoplasmic/membranous")
<br><b>Adenocarcinoma, NOS</b> (M-81403)<br><br><b>Antibody staining:</b> High<br><b>Intensity:</b> Strong<br><b>Quantity:</b> >75%<br><b>Location:</b> Cytoplasmic/membranous")
<br><b>Adenocarcinoma, NOS</b> (M-81403)<br><br><b>Antibody staining:</b> High<br><b>Intensity:</b> Strong<br><b>Quantity:</b> >75%<br><b>Location:</b> Cytoplasmic/membranous")
<br><b>Adenocarcinoma, NOS</b> (M-81403)<br><br><b>Antibody staining:</b> High<br><b>Intensity:</b> Strong<br><b>Quantity:</b> >75%<br><b>Location:</b> Cytoplasmic/membranous")
<br><b>Adenocarcinoma, NOS</b> (M-81403)<br><br><b>Antibody staining:</b> High<br><b>Intensity:</b> Strong<br><b>Quantity:</b> >75%<br><b>Location:</b> Cytoplasmic/membranous")
<br><b>Adenocarcinoma, NOS</b> (M-81403)<br><br><b>Antibody staining:</b> High<br><b>Intensity:</b> Strong<br><b>Quantity:</b> >75%<br><b>Location:</b> Cytoplasmic/membranous")
<br><b>Adenocarcinoma, NOS</b> (M-81403)<br><br><b>Antibody staining:</b> High<br><b>Intensity:</b> Strong<br><b>Quantity:</b> >75%<br><b>Location:</b> Cytoplasmic/membranous")
<br><b>Adenocarcinoma, NOS</b> (M-81403)<br><br><b>Antibody staining:</b> High<br><b>Intensity:</b> Strong<br><b>Quantity:</b> >75%<br><b>Location:</b> Cytoplasmic/membranous")
<br><b>Adenocarcinoma, uncertain malignant potential</b> (M-81401)<br><br><b>Antibody staining:</b> High<br><b>Intensity:</b> Strong<br><b>Quantity:</b> >75%<br><b>Location:</b> Cytoplasmic/membranous")
<br><b>Adenocarcinoma, uncertain malignant potential</b> (M-81401)<br><br><b>Antibody staining:</b> High<br><b>Intensity:</b> Strong<br><b>Quantity:</b> >75%<br><b>Location:</b> Cytoplasmic/membranous")




