We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
General description of the gene and the encoded protein(s) using information from HGNC and Ensembl, as well as predictions made by the Human Protein Atlas project.
Human gene namei
Official gene symbol, which is typically a short form of the gene name, according to HGNC.
All transcripts of all genes have been analyzed regarding the location(s) of corresponding protein based on prediction methods for signal peptides and transmembrane regions.
Genes with at least one transcript predicted to encode a secreted protein, according to prediction methods or to UniProt location data, have been further annotated and classified with the aim to determine if the corresponding protein(s) are secreted or actually retained in intracellular locations or membrane-attached.
Remaining genes, with no transcript predicted to encode a secreted protein, will be assigned the prediction-based location(s).
The annotated location overrules the predicted location, so that a gene encoding a predicted secreted protein that has been annotated as intracellular will have intracellular as the final location.
The RNA data was used to cluster genes according to their expression across tissues. Clusters contain genes that have similar expression patterns, and each cluster has been manually annotated to describe common features in terms of function and specificity.
Neurons - Mixed function (mainly)
Tissue specificityi
The RNA specificity category is based on mRNA expression levels in the consensus dataset which is calculated from the RNA expression levels in samples from HPA and GTEX. The categories include: tissue enriched, group enriched, tissue enhanced, low tissue specificity and not detected.
The tissue data for RNA expression obtained through Cap Analysis of Gene Expression (CAGE) generated by the FANTOM project are reported as Scaled Tags Per Million. Color-coding is based on tissue groups, each consisting of tissues with functional features in common. To access sample data, click on tissue name or bar.
The regional specificity category is based on mRNA expression levels in the analysed brain samples, grouped into 13 main brain regions and calculated for the three different species. All brain expression profiles are based on data from HPA. The specificity categories include: regionally enriched, group enriched, regionally enhanced, low regional specificity and not detected. The classification rules are the same used for the tissue specificity category.
Low region specificity
Low region specificity
Low region specificity
Tau specificity scorei
Tau specificity score is a numerical indicator of the specificity of the gene expression across cells or tissues. The value ranges from 0 and 1, where 0 indicates identical expression across all cells/tissue types, while 1 indicates expression in a single cell/tissue type.
0.07
0.10
0.05
Regional distributioni
The regional distribution category is based on mRNA expression detected above cut off or not in the analysed brain samples, grouped into 13 main brain regions and calculated for the three different species. Brain expression for all species is based on data from HPA. The distribution categories include: detected in all, detected in many, detected in some, detected in single and not detected. The classification rules are the same used for the tissue distribution category.
Detected in all
Detected in all
Detected in all
Cellular locationi
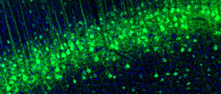
Manually selected location of the protein positivity, observed by immunofluorescence staining in mouse brain.
Soma and synapse in neurons.
Protein profilei
A summary of the overall protein localization across the analyzed brain tissue. The summary is based on manual annotation Mouse brain profiles includes a more extensive summary of the protein location while summaries with only human protein profile are more limited and are based on imported data/images from the Tissue resource stainings that has undergone an additional brain-centric curation step.
Strong intensity diffuse/synaptic-type immunoreactivity is noticed throughout the entire brain gray matter. In addition, neuronal cell bodies are immunoreactive in the oculomotorius nucleus and in the locus coeruleus.
BRAIN RNA EXPRESSIONi
The data is based on "bulk" RNAseq of micro-punched brain areas and brain nuclei. The size of the punches was adjusted to the size of the brain region to obtain detailed protein expression information on distinct cell populations. Multiple samples were collected from the same donors allowing a mild normalization and correction for individual variation. Data is expressed as normalized tags per million (nTPM). Protein expression data is organized on the 13 main brain structures representing the maximum value found in any of the brain areas that make up that structure. A detailed overview of the expression in any of the brain areas is summarized in bar plots for each brain structure.
Normalized RNA expression levels (nTPM) shown for the 13 brain regions. Color coding is based on brain region and the bar shows the highest expression among the subregions included. To access sample data, click on region name or bar.
Read more about normalized expression levels here.
CTX
HPF
AMY
BG
TH
HY
MB
CB
P
M
SC
WM
CP
Cerebral cortex
Additional Prefrontal Cortex dataset
Hippocampal formation
Amygdala
Basal ganglia
Thalamus
Hypothalamus
Midbrain
Cerebellum
Pons
Medulla oblongata
Spinal cord
White matter
Choroid plexus
STEREO-SEQi
The left image is based on imputation of transcript location based on single cell co-expression. Only locations that are supported by neighboring transcripts are shown. All pixels are allocated to a cell-type mask indicated by color. It should be noted that these are predictions on transcript location and not real detection of transcripts.
The bar plot on the right reveals the counts per million of that transcript in each cell-type mask. This is calculated by counting all transcripts for all cell-type masks and for unassigned spots (noise). For each protein coding transcript the unassigned counts are subtracted from the counts in each cell-type. The noise corrected counts are used to calculate counts per million that is plotted as Enrichment change. Note that this is based on real measurements of counts.
This is data based on "bulk" RNAseq (GTEx) and Cap Analysis of gene expression. Protein expression is is expressed as normalized tags per million (nTPM).
GTEX AND FANTOM HUMAN BRAIN
GTEx Human brain RNA-Seq dataseti
GTEx dataset RNA-seq tissue data generated by the Genotype-Tissue Expression (GTEx) project is reported as mean nTPM, corresponding to mean values of the different individual samples for respective subregion. Highest expression among the subregions represents the brain region. To access sample data, click on region name or bar.
The GTEx RNA-seq assay is described in detail here.
FANTOM5 Human brain CAGE dataseti
FANTOM5 dataset Tissue data for RNA expression obtained through Cap Analysis of Gene Expression (CAGE) generated by the FANTOM5 project are reported as Scaled Tags Per Million. To access sample data, click on region name or bar.
HPA Pig dataset HPA RNA-seq tissue data is reported as mean nTPM (normalized expression) for each of the brain regions analyzed in pig. The detailed pages (reached when clicking a bar or regional name) show nTPM values at the individual sample level. To access sample data, click on region name or bar.
The HPA RNA-seq assay is described in detail here.
The pig brain transcriptomics project is a collaborative project between human protein atlas and the Lars Bolund institute of regenerative Medicine (Dr. Yonglun Luo), BGI-Qingdao, China.
HPA Mouse dataset HPA RNA-seq tissue data is reported as mean nTPM (normalized expression) for each of the brain regions analyzed in mouse. The detailed pages (reached when clicking a bar or regional name) show nTPM values at the individual sample level. To access sample data, click on region name or bar.
The HPA RNA-seq assay is described in detail here.
Mouse brain protein profiles are based in serial sections of the adult mouse brain analyzed by immunofluorescence. The staining intensity and regional distribution is manually summarized.
Protein profile
Strong intensity diffuse/synaptic-type immunoreactivity is noticed throughout the entire brain gray matter. In addition, neuronal cell bodies are immunoreactive in the oculomotorius nucleus and in the locus coeruleus.
Cellular location
Soma and synapse in neurons.
Protein profile reliability
Supported based on CAB079051
HPA Mouse staining intesityi
The score is based on the relative fluorescence, defined as intensity of the annotated region relative to the intensity of the region with highest intensity.
The digitalized images are processed (axel-adjusted and tissue edges defined) and regions of interest (ROIs) are then marked. These ROIs are then used for image analysis and the relative fluorescence intensity is listed for each region. For more data and regional information click on the bar or the regional name.
Selected annotations
Thalamus
Hippocampal formation
Pons
Hippocampal formation
FRONTBACK
EXPRESSION CLUSTERING & CORRELATIONi
The RNA data was used to cluster genes according to their expression across samples. The resulting clusters have been manually annotated to describe common features in terms of function and specificity. The annotation of the cluster is displayed together with a confidence score of the gene's assignment to the cluster. The confidence is calculated as the fraction of times the gene was assigned to this cluster in repeated calculations and is reported between 0 to 1, where 1 is the highest possible confidence. The clustering results are shown in a UMAP, where the cluster this gene was assigned to is highlighted as a colored area in which most of the cluster genes reside. A table shows the 15 most similar genes in terms of expression profile.
TPRG1L is part of cluster 24Neurons - Mixed function with confidencei
Confidence is the fraction of times a gene was assigned to the cluster in repeated clustering, and therefore reflects how strongly associated it is to the cluster. A confidence of 1 indicates that the gene was assigned to this cluster in all repeated clusterings.
Correlation between the selected gene and neighboring gene. Correlation is calculated as Spearman correlation in PCA space based on the RNA-seq expression data.
Clusteri
ID of the expression cluster of the neighboring gene.
 The Human Protein Atlas project is funded
The Human Protein Atlas project is funded